



什么是染色体异常遗传病
染色体异常遗传病(简称“染色体病”),指染色体的数目异常和形态结构畸变,可以发于每一条染色体上。
染色体异常遗传病在自发性流产、死胎、早夭中占50%以上,新生儿中发病率约点1%,是性发育异常及男女不孕症、不育症的重要原因,也是先天性心脏病、智能发育不全等的重要原因之一。随着染色体分带技术、PCR技术、DNA检测技术等的发展,对染色体畸变与疾病关系的认识日益加深,染色体病日趋增多。
综合许多国家的资料,大约有15%的妊娠发生流产,而其中一半为染色体异常所致,即约为5%-8%的胚胎有染色体异常。不过在出生前,90%以上已有自然流产或死产。流产愈早,有染色体异常的频率愈高。
分类:
常染色体异常
性染色体异常
三体综合征(常染色体异常)
先天愚型
先天愚型是最主要的染色体疾病。英国医生Langdon Down 首先描述,故称为 Down综合征(Down sydrome)。1959年,法国细胞遗传学家Lejeune证实此病的病因是多了一个小的G组染色体(后来确定为21号),故此病又称为21三体综合征。 Lejeune的发现开创了医学遗传学的一个重要分支――临床细胞遗传学。
发病率
新生儿中21三体综合征的发病率约为1/800或1.25%,但男性患儿多于女性。母亲年龄是影响发病率的重要因素。根据国外资料,如果一般人出生时的母亲年龄平均28.2岁,则本病患儿母亲平均年龄为34.4岁,如母龄20岁后为1:2000,35岁后为1:300,40岁后为1:100,45岁后升至1:50。
临床表现
先天愚型患儿出生时体重和身长偏低,肌张力低下,突出的是颅面部畸形,头颅小而圆,枕部扁平,脸圆而扁平,鼻扁平,脸裂细且上外倾斜,眼距过宽,内眦赘皮明显,常有斜视,虹膜时有白斑点,常见晶状体混浊,嘴小唇厚,舌大外伸(伸舌样痴呆之名由此而来),耳小,耳位低,耳廓畸形,颈背部短而宽,有多余的皮肤,由于软骨发育差,患者四肢较短,手宽而肥,通贯掌,指短,第5指常内弯,短小或缺小指中节,皮纹也有一定的特点(参阅第十章),腹肌张力低下而膨胀,故常有腹直肌分离或脐疝,约1/2以上的患者有先天性心脏病,主要是室间隔缺损、房室道连通、和运脉导管未闭,消化道的畸形如十二指肠的狭窄、巨结肠、直肠脱垂及肛门闭锁等也偶尔可见。
在男性常有隐睾,睾丸有生精过程,但精子常减少,性欲下降,尚未见有生育者。女性患者通常无月经,但有少数能妊娠和生育。精神发育迟滞或智力低下(mental retardation,MR)是本病最突出最严重的表现,但其程度在各患者不完全相同,智商通常在25-50之间,高于50的很少。行为、动作倾向于定型化,抽象思维能力受损最大。
实验室检查
过氧化物岐化酶(SOD-1)活性可增高50%,该酶基因定位21q22,即具有基因剂量效应。此外,患者对阿托品特别敏感,患者乙酰胆碱缺陷或许可以解释智力低下、应变力差、便秘等症状,而免疫功能失调,如淋巴细胞和丙球蛋白减少则是患儿易感染的原因。
核型
核型可分为三型,各型的比例是:典型的(游离型),即47,+21占95%;嵌合型即46/47,+21占1%-2%;易位型占3%-4%。游离型全身体细胞均多一条21号染色体,临床症状典型而且显著。由于嵌合型通常具有两个细胞系,其症状表现取决于异常细胞所占的比例,故差异很大,但一般较典型者为轻。如果三体细胞很少,则表现与正常人无异。易位型的核型有多种,最常见的是Dq21q,占全部易位型的 54.2%,其次是21qGq,占40.9%,其它易位型5%。一般说来,易位型的症状比典型的要轻些,在Dq21q中,最常见的是14q21q,占Dq21q的58.5%,其次为13q21q,占22%,而15q21q占19.5%。21qGq易位中,21q21q占83.3%,而21q22q仅占16.6%。无论体积易位,患者虽然只有46条体,但因一条21号易位到了另一条D组或G组染色体上,加上正常的两条21号,仍多了一条额外的21号长臂,而决定本病的关键区带为21号长臂,故临床上仍表现出21三体的症状。
遗传学
典型的21三体几乎都是新发生(de novo)的,与父母的核型无关,经是减数分裂时不分离的结果。不分主离常发生在母方生殖细胞,约占病例数的95%,另5%见于父方,而且主要发生在第一次减数分裂。典型的21三体只有极少一部分是遗传的,即母亲是本病患者。此外,不能排除某些表型正常的母亲实际是21三体细胞较少的嵌合体,因而她们的子女有可能获得额外的21号染色体。男性患者不能生育,没有遗传给下一代的问题。
嵌合型患者有两个或两个以上的细胞系。它们是合子后(post-zygotic)有丝分裂不分离的结果。如果第一次卵裂时发生不分离,就会产生47,+21和45,-21两个细胞系。而后一种细胞是很维存活的。因此,导致嵌合体的不分离多半发生在以后的某次有丝分裂,所有嵌合体内都有正常的细胞系。
易位型的21三体征患者细胞中有一条易位的染色体,后者通常由一条D组或G组染色体与21号染色体长臂通过着丝粒融合(罗氏易位)而成。Dq21q易位中,55%是新发生的,45%是由于双亲之一有平衡易位。21qGq 易位几乎全部(96%)是新发生的,由遗传而来的仅占4%。
各种易位的遗传后果不同。Dq21q平衡易位的携带者通过减数分裂可以形成6种配子,而受精后除不能发育者外,可以产生正常胎儿、易位型三体患儿和平衡易位携带者三种胎儿(图2-12)。因此,检出平衡易位携带者的双亲具有重要意义。
21qGq易位中,21q22q和21q21q易位的遗传学意义不完全相同。如果双亲之一为21q21平衡易位携带者,就没有可能娩出表型正常的胎儿,因为他们只能产生三体或单体的合子。21q22q易位的遗传后果与Dq22q相似,只是前者更多通过父亲传递而后者多由母亲传递得来。
遗传咨询
各种类型再发风险不同。对游离型的21三体性,再发风险率与年龄特异风险率相近(即35岁以下约为0.5%,35%岁以上约为1%)。然而,如双亲(通常是母亲)之一嵌合体则风险会增加。总之,已产生一个21三体患儿,其再发风险0.5%,而染色体畸变的总风险为1.2%,即不排除再生产其它染色体异常的患儿。
易位型有大约1/2的病例是新发生的,另1/2是双亲的之一平衡易位引起的。前者复发风险很小,而平衡易位导致的再发风险则可以根据经验估计。理论上如双亲之一为平衡易位携带者,产出患儿风险为33.3%。但实际风险低得多,这与双亲哪一方为携带者有关。Dq21q易位携带者若是母亲,生产患儿的风险为10%-15%;如为父亲,则风险为5%或更小。21q21q易位的情况与之大体相同,但易位染色体由父方传递的百分比较D/G易位要多,风险率在10%以下。21q21q平衡易位携带者的后代100%均是三体征患儿,携带者不宜生育。
预后
患者平均寿命只有16.2岁。50%的患儿在5岁以前死亡。只有8%的患者超过40岁,2.6%超过50岁。根据四川省的资料,人群中患病率仅约为再生时的1/10。
13三体综合征
1960年Patau首先描述本病,故又称为Patau综合征。新生儿中的发病率约为1:25 000,女性明显多于男性。
临床表现
患儿的畸形和临床表现要比21三体性严重得多。颅面的畸形包括小头,前额、前脑发育缺陷,眼球小,常有虹膜缺损,鼻宽而扁平,2/3患儿有上唇裂,并常有腭裂,耳位低,耳廓畸形,颌小,其它常见多指(趾),手指相盖叠,足跟向后突出及足掌中凸,形成所谓摇椅底足。男性常有阴囊畸形和隐睾,女性则有阴蒂肥大,双阴道,双角子宫等。脑和内脏的畸形非常普遍,如无嗅脑,心室或心房间隔缺损、动脉导管未闭,多囊肾、肾盂积水等,由于内耳螺旋器缺损造成耳聋。
智力发育障碍见于所有的患者,而且程度严重,存活较久的患儿还有癫痫样发作,肌张功力低下等。
细胞遗传学及遗传咨询
80%的病例为游离型13三体,核型为46,XX(或XY),+13,其余的则为嵌合型或易位型。嵌合型一般症状较轻,易位型通常以13和14号罗氏易位居多,患者有一条t(13q14q)易位染色体,核型为46,-14,+t(13q14q),其结果是多了一条13号长臂,当双亲之一是平衡易位携带者时,因为绝大多数异常胎儿均流产死亡,产出患儿的风险不超过5%或1%。如果双亲之一为13q13q易位携带者,由于只能产生三体或单体的合子,流产率达100%。
病因及预后
与13三 体发和表关的因素所知甚少,母亲高龄可能是原因之一,患儿母亲的平均年龄为31.6岁,父亲的平均年龄为34.6岁。此外,有资料表明,79%病例妊娠于寒冷季节(9-2月),45%的患儿在出生后一个月内死亡,90%在6个月内死亡,存活至3岁者少于5%,平均寿命为130天。
18三体综合征
1960年Edward等首先描述,故又称为Edward综合征(Edward syndrome ).18三体性导致严重畸形,在出生后不久死亡。发病率约1:3500-8000新生儿。但在某些地区或季节明显增高,达到1:450-800。患儿中女性:男性比为4:1。
临床表现
患儿出生时体重低,平均仅2243g,发育如早产儿,吸吮差,反应弱,头面部和手足有严重畸形,头长而枕部凸出,面圆,眼距宽,有内赘眦皮,眼球小,角膜混浊,鼻梁细长,嘴小,耳位低,耳廓畸形(动物样耳),小颌(micrognathia),颈短,有多余的皮肤,全身骨骼肌发育异常,胸骨短,骨盆狭窄,脐疝或腹股沟疝,腹直肌分离等。手的畸形非常典型:紧握拳,拇指横盖于其它指上,其它手指互相叠盖,指甲发育不全,手指弓形纹过多,约1/3患者为通贯掌。下肢最突出的是“摇椅底足”,拇趾短,向背侧屈起。外生殖器畸形比较常见的有隐睾或大阴唇和阴蒂发育不良等。95%的病例有先天性心脏病,如室间隔缺损、动脉导管未闭等,这是死亡的重要原因。肾畸形,肾盂积水也很常见。患儿智力有明显缺陷,但因存活时间很短,多数难以测量。
细胞遗传学
80%患者核型为47,XY(或XX),+18;另10%患者为嵌合体,即为46,XY(或XX)/47,XY(或XX),+18;其余为各种易位,主要是18号与D组染色体的易位。双亲是平衡易位携带者而导致18三体征者很少。
预后
患儿大多在2-3个月内死亡,平均存活71天,只有极个别病人超过儿童期。嵌合型的存活期比较长。
其它染色体三体综合征
比较重要的有8号、22号三体综合征等。都伴有明显的发育畸形和智力低下。还有一系列由易位引起染色体部分三体综合征,其临床症状取决于额外染色体片段的性质和大小。染色体部分三体性可分为两大类:一类有某一染色体片段的三体性(重复),同时又伴有其它染色体的异常(如缺失、易位),这一类部分三体性的表型比较复杂,常兼有重复和缺失片段的某些症状;另一类为染色体的某一片段的单纯重复或三体性,这在人类极为少见。
单体及部分单体综合征(常染色体异常)
整条常染色体的丢失通常是致死的,因而极为罕见,但确有小染色体(如21号)完全丢失的报告。由于易位、环形成或缺失导致的染色体部分单体则比较多见。现以5q-综合征,即猫叫综合征为介绍如下。
猫叫综合征(5q-综合征)为最见的缺失综合征,其发病率估计为1:50000,女性多于男性。患婴的哭叫声非常似小猫的咪咪声,故得名。患儿面部情似很机灵,但实则智力低下非常严重(智商常低于20),发育迟滞也很明显。常见的临床表现还有小头、满月脸、眼裂过宽、内眦赘皮、下颌小且后缩(图2-20)。约20%患者有先天性心脏病,主要是室间隔缺损和动脉导管未闭等。
患者的染色体缺失片段大小不一。症状主要由5p15的缺失引起。畸变多数是新发生的。由染色体片段的单纯缺失(包括中间缺失)的占80%,不平衡易位引起的占10%,环状染色体或嵌全体则比较少见,由亲代染色体重排导致的5p-综合征不多见。
性染色体数目异常综合征
Klinefelter综合征
(Klinefelter syndrome)又称为先天性睾丸发育不全或原发小睾丸症。患者性染色体为XXY,即比正常男性多了一条X染色体,因之本亦常称为XXY综合征。
Klinefelter综合征的发病率相当高,男性新生儿中达到1.2‰。根据白种人的资料,身高180cm的男性患病率为1/260,在精神病患者或刑事收容机构中为1/100,在因不育而就诊者中约为1/20。临床表现为睾丸小而质硬,曲细精管萎缩,呈玻璃样变。由于无精子产生,故97%患者不育。患者男性第二性征发育差,有女性化表现,如无胡须,体毛少,阴毛分布如女性,阴茎龟头小等,约25%的患者有乳房发户。患者身材高。四肢长,一部分患者(约1/4)有智力低下,一些患者还有精神异常及患精神分裂症倾向。实验室检查可见雌激素增多,19-黄体酮增高,激素的失调与患者的女性化可能有关。
绝大多数患者的核型为47,XXY。大约有15%患者为两个或更多细胞系的嵌合体,其中常见的为46,XY/47,XXY;46,XY/48,XXXY。额外的X是由于亲代减数分裂时X染色体不分离的结果。
用睾丸酮治疗可以收到一定的效果,它可促使第二性征发良并性病患者的心理状态。
XYY综合征
在男婴中的发生率为1:900。XYY男性的表型的正常的,患者身材高大,常超过180cm,偶尔可见隐睾,睾丸发育不全并有精过程障碍和生育力下降,尿道下裂等,但大多数男性可以生育。XYY个体易于兴奋,易感到欲望不满足,厌学,自我克制力差,易产生攻击性行为。XYY核型是父亲精子形成过程中第二次减数分裂时发生Y染色体不分离的结果。
Turner综合征
又称为45,X或45,XO综合征、女性先天性性腺发育不全或先天性卵巢发育不全综合征。在新生女婴中的发病率约为0.2‰-0.4‰,但在自发流产胚胎中Turner综合征的发生率可高达7.5%.患者表型为女性,身材矮小,智力一般正常,但常低于其同胞,面呈三角形,常有睑下垂及内眦赘皮等,上颌突窄,下颌小且后缩,口角下旋呈鲨鱼样嘴,颈部的发际很低,可一直伸延到肩部,约50%患者有蹼颈,即多余的翼状皮肤,双肩径宽,胸宽平如盾,乳头和乳腺发育差,两乳头距宽,肘外翻在本病十分典型,第四、第五掌骨短而内弯,并常有指甲发育不全。婴儿期脚背有淋巴样肿,十分特殊。泌尿生殖系统的异常主要是卵巢发育差(索状性腺),无滤泡形成,子宫发育不全,常因原发性闭经来就诊。由于卵巢功能低下患者的阴毛稀少,无腋毛,外生殖器幼稚。此外,大约有1/2患者有主动脉狭窄和马蹄肾等畸形。
Turner综合征的核型除典型的45,X(约占55%)外,还有各种嵌合型和结构异常的核型。最常见的是嵌合型46,XX/45,X和46,X,i(Xq)。一般说来,嵌合型的临床表现较轻,而有Y染色体的嵌合型可表现出男性化的特征,身材矮小和其它Turner症状主要是由X短臂单体性决定的,但卵巢发育不全与不育则更多与长臂单体性有关。
Turner综合征的发病机理是双亲配子形成过程中的不分离,其中约75%的染色体丢失发生在父方,约10%的丢失发生在合子后早期卵裂时。
除少数患者由于严重畸形有新生儿期死亡之外,一般均能存活,只是在青春期才被检出。其智力发育障碍也较轻,应用激素在14岁以前开始治疗可以促进第二性征和生殖器官的发育,月经来潮,心理状态改变,但不能促进长高,个别患者可生育。
47,XXX和X女性
本病又称为超雌(superfemale)。发病率约为0.44‰或1/2250。
多数具有三条X染色体的女性无论外形、性功能与生育力都是正常的,只有少数患者有月经减少、继发闭经或过早绝经等现象。大约有2/3病智力稍低,并有患精神病倾向。
除了47,XXX外,一些患者的核型为嵌合型,即47,XXX/46,XX。XXX患者的母亲生育年龄平均约增高4岁。这表明不分离主要发生在母亲一方。少数患者有4条甚至5条X染色体。一般来说,X染色体愈多,智力损害和发育畸形愈严重。
性染色体的结构畸变
X染色体的结构异常
常见的X染色体结构异常有各种缺失、易位和等臂染色体。它们的临床表现多样,主要取决于涉及X染色体上的哪些区段异常,因为不同的区段载有的基因不同,缺失导致的症状也不同。Wyss等曾作一Turner综合征的核型与症状关系图。有助于了解X染色体结构异常的表现。
(1)X短臂缺失(XXp-):Xp远端缺失病人有诸如身材矮小等Turner综合征特征,但性腺功能正常。Xp缺失如包括整个短臂,则患者既有Turner综合征的体征,又有性腺发育不全。X染色体长臂等染色体[X,i,(Xq)]的临床表现与此类似,因为也缺失了整个短臂。
(2)X长臂缺失(XXq-):缺失在q22以远者,一般仅有性腺发育不全,原发闭经,不育,而无其它诸如身材矮小等Turner综合征体征。缺失范围较大,包括长臂近端者,有性腺发育不全外,一些患者还有其客观存在体征。X染色体等臂染色体[Xi(p)]与此类似。Xq中间缺失累及q13-q26者性腺功能正常,但有其它体征,可见中段缺失与Turner体征出现有关。
通常部分缺失`形成环状或等臂染色体的X均选择性地失省事,从而保证有一条正常的X。
(3)易位:当X染色体与常染色体发生平衡易位时,由于基因平衡的保持,一般不会产生症状,此时失活的正常的X染色体。但如平衡易位断点在q12-q26时,有活性的X在该区被分为两部分,就会导致性腺发育异常。此外,如常染色体节段易位到X染色体产生不平衡易位时,多数产生双着丝粒染色体,其表型取决于Xp或Xq上断裂点的位置。
脆性X染色体综合征
本世纪初一些学者注意到智力下患者中逻辑性多于女性。1943年Martin和Bell在一个家系两代人中发现11名逻辑性患者和两名轻度智力低下的女性,认为该家系智力低下是与X连锁的,因此X连锁智力低下又称为Martin-Bell综合征。
1969年Lubs首先在逻辑性智力低下患者及其女性亲属中发现了长臂具有“随体和呈细丝状次缢痕”的X染色体。后来,Sortherland 证明细丝位位于X染色体长臂2区7带(Xq27)。它在低叶酸培养条件下表达,并提出了脆性部位(franile site)的概念。现今人们把在Xq27处有脆性部位的X染色体称为X染色体(fragile X,fra X),而它所导致的疾病称为脆性X染色体综合征。
(1)发病率:本病在逻辑性中的发病率为1/1000~1/1500,仅次于先天愚型。在所有逻辑性智力低下患者约10%~20~为本病所引起。
(2)临床表现:主要表现为中度到重度的智力低下,其它常见的特征尚有身长和体重超过正常儿,发育快,前额突出,面中部发育不全,下颌大而前突,大耳,高腭弓,唇厚,下唇突出,另一个重要的表现是大睾丸症。一些患者还有多动症,攻击性行为或孤癖症。20%患者有癫痫发作。过去曾认为由于女性有两条X染色体,因此女性携带者不会发病,但由于两条X染色体中有一条失活,女性杂合子中约1/3可有轻度智力低下。
(3)发病的分子机理:现今在X脆性部们已发现了致病基因FMR-1,它含有(CGC)n 三核甘酸重复序列,后者在正常人约为30拷贝,而在正常男性传递者和女性携带者增多到150~500bp,称为小插入,相邻的Cpg 岛未被甲基化,这种前突变(premutation )无或只有轻微症状。女性携带者的CGG区不稳定,在向受累后代传递过程中扩增,以致在男性患者和脆性部位高表达的女性达到1000~3000bp,相邻的CpG岛也被甲基化。这种全突变(full mutation)可关闭相邻基因的表达,从而出现临床症状。由前突变转化为完全突变只发生母亲向后代传递过程中。根据对脆性部位DNA序列的了解,现已可用RFLP连锁分析、DNA杂交分析、PCR扩增等方法来检出致病基因。
(4)治疗:Lejeune 认为叶酸缺乏是Fra X综合征时智力低下的原因,他用大剂量叶酸治疗患者获得了良好的效果,但其他作者未能证实叶酸的疗效。新近一些作者认为中枢神经兴奋剂疗效较好,但副作用大。其它有用可乐定(clonidine)、心得安者,据称可减轻多动症。
Y染色体及其结构异常
Y染色体用荧光染色时,因长臂未端有宽阔的极明亮的荧光带,易与21、22号区别。Y染色体长臂的多态性非常明显,存在种族差异,大Y在中国人和日本人中的比例较高。
Y染色体的数目异常
包括前已述及的XYY以及超Y综合征(XXYY等)。Y染色体的结构异常包括Y的长臂或短臂缺失、等臂染色体i(Yq)和i(Yp)、环状染色体和双着丝粒染色体(为两条Y的短臂相连或两条Y的长臂相融合)、倒位和各种涉及Y的易位(即Y与常染色体,Y与X染色体的易位等)。此外性反转综合征46,XX男性是Yq上的SRY基因易位到一条X染色体所致,而46,XY女性是SRY基因缺失或突变的结果。
什么是无创产前检测?
无创产前检测是一项高度准确的筛查技术,与传统入侵性检验方法不同,传统入侵性如绒毛活检、羊膜穿刺等检查,存在流产风险,医生指出,无创筛查技术只需抽取少量孕妇血液,无流产风险,而且准确性高于99%,大大减少了误诊的可能性。
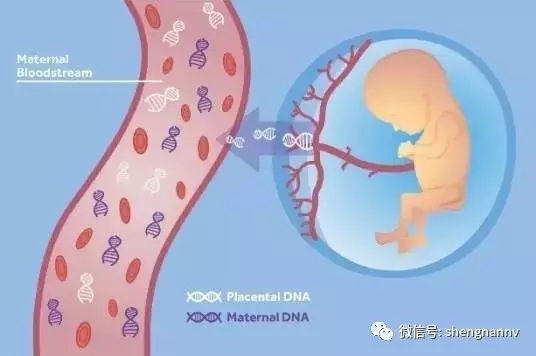
透过无创产检,准妈妈可以取得哪些数据?
产前筛检可测试到婴儿染色体的特定异常情况。人体共有 23 对 (每对两条,合共 46 条) 染色体。前 22 对染色体依次命名为 1 至 22 号染色体,最后一对染色体会决定胎儿性别。女性有两条 X 染色体,男性则会有一条 X 和 一条 Y 染色体。任何染色体的多余或缺失都会引起健康及发育问题。
三体症是指某一染色体多出了一个复本 — 正常情况下只有 2 条的染色体,在三体症情况下则变为 3 条。
单染色体症是指某一染色体缺少了一个复本— 正常情况下应有 2 条的染色体,在单染色体症情况下则只有 1 条。
三倍体是指所有染色体均有 3条的异常情况。

安心微信客服号

安心微信客服号